|
| |
|
|
 |
|
Bouffée délirante : définition, symptômes, traitements |
|
|
| |
|
| |

SANTÉ MALADIE
Bouffée délirante : définition, symptômes, traitements
Par Sciences et Avenir le 12.02.2014 à 11h23, mis à jour le 18.01.2023 à 16h33
Lecture 3 min.
La bouffée délirante se manifeste de manière brutale et subite : la personne a alors un comportement très inhabituel avec des changements rapides d’humeur et de posture. Cet épisode a une durée variable de quelques heures à quelques semaines et se reproduit rarement.
Tout le monde, sans trouble psychique particulier, peut développer un jour une bouffée délirante mais elle touche en général des personnes jeunes (18 à 30 ans).
© CREATIVE COMMONS
Définition et symptômes de la bouffée délirante
La bouffée délirante (BD) est un état psychotique aigu reconnue uniquement en France. Il revient à Valentin Magnan de l’avoir décrite en 1866. La bouffée délirante se traduit par un état psycho-pathologique aigu avec l’apparition brutale d’un délire très riche en matière d’expressions et de thèmes. Le sujet est alors en totale adhésion avec ce qu’il vit.
C’est un bouleversement psychique avec au moins trois de ces critères :
- un changement soudain d’une émotion à une autre, comme de la colère à l’angoisse ;
- un changement d’humeur : de l’euphorie à la dépression ;
- un changement de comportement psychomoteur : de l’agitation à la prostration ;
- une dépersonnalisation ;
- des hallucinations avec des thèmes comme la persécution, la jalousie ou encore la mégalomanie.
Tout le monde, sans trouble psychique particulier, peut développer un jour une bouffée délirante mais elle touche en général des personnes jeunes (18 à 30 ans). Certaines substances peuvent déclencher cet épisode d’hallucinations et de délires. Il a également été constaté une fragilité psychologique des personnes atteintes. Les délires peuvent revêtir plusieurs formes comme des hallucinations ou des voix intérieures. Les phases euphoriques puis dépressives se succèdent avec une modification de la qualité du sommeil. Après une adhésion totale à ses délires, le patient prend conscience de son état, ce qui le laisse dans un état de perplexité, souvent dépressif.
Des nouvelles techniques permettent de détecter les risques de développer une psychose.
Evolution de la bouffée délirante
Ces crises, qui durent des heures, des jours ou des semaines, peuvent se résorber lentement ou brutalement. Une fois passé, dans un tiers des cas, le trouble ne se renouvelle pas. Il évolue rarement vers d’autres maladies psychiques telles qu’une psychose maniaco-dépressive. Néanmoins, les épisodes de bouffée délirante peuvent apparaître chez des sujets et devenir chroniques. Enfin, cela peut aussi être un symptôme de la schizophrénie.
Les causes de la bouffée délirante
La bouffée délirante peut avoir des origines multiples :
- un contexte relationnel difficile (affectif, familial, professionnel) qui provoque anxiété et angoisse ;
- un surmenage, un manque de sommeil ;
- la consommation régulière de drogues.
Traitement et prise en charge de la bouffée délirante
Trois étapes s’imposent :
- une hospitalisation psychiatrique, souvent en urgence, même sans l’accord de l’intéressé, est nécessaire pour connaître et traiter la cause des symptômes ;
- un traitement médical (neuroleptiques) est ensuite prescrit ;
- enfin, même si la crise n’est apparue qu’une seule fois, il faut consulter un spécialiste pour dépister l’évolution et approfondir les déclencheurs et les causes profondes du problème. Une psychothérapie de soutien peut être nécessaire.
DOCUMENT sciences et avenir.fr LIEN |
| |
|
| |
|
|
|
Une nouvelle canalopathie cérébrale associant déficience intellectuelle et mouvements anormaux |
|
|
| |
|
| |
Une nouvelle canalopathie cérébrale associant déficience intellectuelle et mouvements anormaux
27 NOV 2020 | PAR INSERM (SALLE DE PRESSE) | NEUROSCIENCES, SCIENCES COGNITIVES, NEUROLOGIE, PSYCHIATRIE
Brain scan, X-ray © Fotolia
Les dysfonctionnements des canaux ioniques – ou canalopathies – dans le cerveau sont aujourd’hui associés à plus de 30 maladies neurologiques comme l’épilepsie ou encore les ataxies cérébelleuses. Structures situées sur la membrane des cellules permettant le passage d’ions (par exemple les ions sodium et potassium) entre l’intérieur d’une cellule et son environnement extérieur (milieu extracellulaire), ces canaux permettent notamment de générer et contrôler les potentiels d’action dans les neurones. Une étude menée à l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS) a permis d’identifier une nouvelle canalopathie cérébrale ayant pour origine des mutations dominantes du gène KCNN2, codant pour le canal ionique SK2. Les résultats ont été publiés dans Brain le 27 novembre 2020.
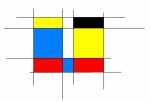
Les variants pathogéniques du gène KCNN2 identifiés chez les patients et leur localisation sur la structure protéique du canal SK2.
Les variant en rouge sont des variants pathogènes tronquant (introduisant un codon stop dans la séquence protéique). Les variants en noirs sont les variants pathogènes faux-sens associés à une perte de fonction. Le variant en gris a été classé de signification inconnue car le canal avec ce variant n’a pas montré de déficit particulier en électrophysiologie.
Le Dr Fanny Mochel, généticienne au sein du département de génétique de l’hôpital de la Pitié-Salpêtrière AP-HP et chercheuse à l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS) et le Pr Christel Depienne, généticienne à l’institut de génétique humaine de l’Hôpital Universitaire d’Essen (Allemagne) et également chercheuse à l’Institut du cerveau ont identifié un nouveau syndrome associé à des mutations du canal SK2. L’étude publiée dans la revue scientifique Brain porte sur 10 patients, 6 hommes et 4 femmes âgés de 2 à 60 ans présentant des retards intellectuels plus ou moins sévères associés, pour certains, à des troubles du spectre autistique ou des épisodes psychotiques. Ces troubles cognitifs sont dans tous les cas associés à des tremblements, à des symptômes d’ataxie cérébelleuse ou encore à des mouvements anormaux.
Grâce à une collaboration avec Agnès Rastetter de la plateforme de génotypage/séquençage de l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS), le génome d’un premier patient recruté à la Pitié-Salpêtrière a été analysé à la recherche de mutations génétiques à l’origine de ce syndrome. Cette analyse a mis en évidence une mutation du gène KCNN2 interrompant sa séquence codante, absente des parents du patient (mutation de novo). L’imagerie cérébrale par IRM (imagerie par résonance magnétique) chez ce patient a mis en évidence des anomalies de structure et d’intégrité de la substance blanche du cerveau, c’est-à-dire la gaine cérébrale protectrice des axones des neurones.
Par ailleurs, une collaboration internationale a permis aux chercheurs d’identifier 9 autres patients avec mutations du gène KCNN2. La majorité de ces mutations étaient survenues de novo tandis qu’une mutation était transmise dans une forme familiale du même syndrome.
Enfin, en travaillant conjointement avec Carine Dalle de la plateforme d’exploration cellulaire d’électrophysiologie de l’Institut du cerveau, les équipes des Dr Mochel et Depienne ont montré un rôle délétère de ces mutations sur la fonction du canal SK2, c’est-à-dire une perte de fonction entrainant un dysfonctionnement du canal ionique SK2 et donc une perte de régulation du potentiel d’action, support du message nerveux.
Les résultats de cette nouvelle étude ont permis d’identifier une nouvelle canalopathie cérébrale ayant pour origine des mutations dominantes du gène KCNN2, codant pour le canal ionique SK2. Ce nouveau syndrome se caractérise par la présence, d’une part, de symptômes cognitifs, en particulier une déficience intellectuelle et, d’autre part, de symptômes moteurs tels que des mouvements anormaux.
Cette nouvelle pathologie, dont on connaît maintenant la cause, est très hétérogène d’un point de vue des symptômes et nécessite une prise en charge multidisciplinaire à la frontière entre la génétique, pour la recherche des mutations du gène KCNN2, la neuropédiatrie et la neurologie pour la prise en charge des manifestations cognitives et motrices des patients.
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Une nouvelle piste pour contrer le choc septique 08 SEP 2017 | PAR INSERM (SALLE DE PRESSE) | |
|
|
| |
|
| |

Une nouvelle piste pour contrer le choc septique
08 SEP 2017 | PAR INSERM (SALLE DE PRESSE) |
PHYSIOPATHOLOGIE, MÉTABOLISME, NUTRITION
Des chercheurs de l’Inserm ont réussi à produire une protéine humaine en laboratoire et à l’utiliser contre les infections bactériennes et comme traitement contre le choc septique. Le choc septique est une réponse inflammatoire généralisée de l’organisme associée à une infection grave. Une fois le stade inflammatoire critique atteint, le pronostic vital des individus est sérieusement engagé. Une personne dans le monde en meurt toutes les 3 à 4 secondes. Ces travaux publiés dans la revue Scientific Reports sont donc une piste sérieuse contre cette infection qui reste aujourd’hui une urgence médicale.
Dans les pays industrialisés, on dénombre 377 cas de sepsis pour 100 000 habitants. Chaque année, le sepsis tue 6 millions de nourrissons. En France, la mortalité des patients atteints d’un sepsis est de 27 %, mais la mortalité de la forme la plus grave peut atteindre 50 %. Les projections suggèrent un doublement du nombre de cas d’ici cinquante ans, s’expliquant notamment par le vieillissement de la population (source : Institut Pasteur). La situation est telle au plan mondial qu’en mai 2017, lors d’une réunion de l’OMS qui s’est tenue à Genève, ses responsables ont pris la décision de reconnaître la septicémie comme un problème de santé publique majeur. La prochaine journée mondiale contre le sepsis, sous l’égide de la Global Sepsis Alliance, se tiendra le 13 septembre 2017.
Dans la plupart des cas, il s’agit d’une infection par des bactéries à Gram négatif présentes naturellement dans l’organisme (la plupart du temps dans l’intestin) qui deviennent toxiques chez des individus fragilisés. La partie toxique de la bactérie se trouve sur leur paroi sous la forme d’un complexe lipo-saccharidique. On parle alors d’endotoxines.
La particularité chimique de ces endotoxines est le point de départ de l’étude menée par les chercheurs de l’Inserm. En effet, de précédentes études ont montré qu’une protéine baptisée PLTP (pour plasma phospholipid transfer protein) avait la faculté de se lier aux endotoxines situées sur la paroi externe des bactéries voire de les transporter vers le foie. En cas d’infection, cette protéine semblait donc pouvoir jouer un rôle dans l’élimination des endotoxines.
Pour vérifier cette hypothèse, l’équipe de recherche, grâce à une collaboration avec une équipe américaine, a étudié un modèle de souris génétiquement modifiée dont la particularité était de ne plus exprimer le gène de la PLTP. En injectant à ces souris des endotoxines bactériennes, les chercheurs ont observé que les animaux meurent sans pouvoir contrer l’infection générée. D’où leur hypothèse que la PLTP présente un intérêt, jusqu’alors inconnu et peut-être majeur, dans le domaine de l’immunité innée.
Tout l’enjeu pour les chercheurs a été ensuite de pouvoir disposer de cette protéine PLTP humaine en quantité suffisante afin de procéder à des essais thérapeutiques visant à montrer sa capacité à contrecarrer les effets de ces endotoxines. Ils se sont alors tournés vers l’Unité mixte de recherche « Biologie du Développement et de la Reproduction » de l’Inra ayant les compétences pour produire la protéine dans le lait de lapines transgéniques.
Une fois cette production obtenue, les chercheurs ont testé la capacité de la PLTP à combattre la réponse inflammatoire chez des souris souffrant de sepsis. D’assez faibles quantités de PLTP ont suffi à améliorer considérablement l’état de santé de ces souris. « Mais, notre objectif ultime était de comprendre comment tout ceci fonctionne » résume Laurent Lagrost.
Poursuivant leurs travaux, les chercheurs ont mis en évidence que la PLTP est capable de bloquer la prolifération des bactéries, en fragilisant leur paroi. Ils observent également que cette protéine qu’est la PLTP, outre la capacité qu’elle a de neutraliser l’activité de ces fameuses endotoxines peut aussi les désagréger avant de les transférer aux lipoprotéines. De simples transporteurs de cholestérol, celles-ci se mutent en véhicule de secours pour convoyer les endotoxines jusqu’au foie et permettre leur élimination par voie biliaire.
« Tant que l’on n’a pas neutralisé de manière endogène, dans l’organisme de l’individu, les endotoxines bactériennes qui vont être responsables de la réponse inflammatoire et de toute la cascade d’effets délétères que cela va entraîner, on ne résout pas définitivement le problème. Or il apparaît que la PLTP, elle, parvient à neutraliser ces endotoxines et à détoxifier le sang, du moins chez les souris » concluent les chercheurs.
Cette démarche s’inscrit dans un concept original de bio-mimétisme où « plus on copie la nature, plus on se rapproche de la vérité », ajoutent-ils.
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Une nouvelle molécule gélifiante pour la culture de neurones en 3D |
|
|
| |
|
| |
Une nouvelle molécule gélifiante pour la culture de neurones en 3D
14 MAI 2018 | PAR INSERM (SALLE DE PRESSE) | TECHNOLOGIE POUR LA SANTE
Une équipe pluridisciplinaire de chercheurs du CNRS, de l’Inserm et de l’Université Toulouse III – Paul Sabatier a mis au point un hydrogel permettant de cultiver des cellules souches neurales, les faire se développer et se différencier. Ce biomatériau pourrait apporter de nouvelles perspectives pour l’élaboration de modèles cellulaires du tissu cérébral in vitro ou la reconstruction tissulaire in vivo. Ces travaux sont publiés dans la revue ACS Applied Materials & Interfaces le 14 mai 2018.
Bien que la culture de cellules soit aujourd’hui bien maîtrisée sur une surface en deux dimensions, cela n’est pas représentatif de l’environnement réel des cellules dans un organisme vivant. En effet, dans le tissu cérébral, les cellules sont organisées et interagissent en trois dimensions dans une structure souple. Ainsi, l’objectif principal pour les chercheurs était d’imiter au mieux ce tissu. Ils ont donc mis au point un hydrogel répondant à des critères de perméabilité, de rigidité et de biocompatibilité adaptés et sur lequel ils ont cultivé des cellules souches neurales humaines 1 .
La N-heptyl-galactonamide est une molécule nouvellement synthétisée par ces scientifiques et fait partie d’une famille de gélifiants habituellement connue pour donner des gels instables. Biocompatible, de structure très simple et rapide à produire, cette molécule présente de nombreux avantages. En travaillant sur les paramètres de formation du gel, les chercheurs des laboratoires Interactions moléculaires et réactivité chimique et photochimique (CNRS/Université Toulouse III-Paul Sabatier), Toulouse Neuro Imaging Center (Inserm/Université Toulouse III-Paul Sabatier) et du Laboratoire d’analyse et d’architecture des systèmes du CNRS ont obtenu un hydrogel stable, très peu dense et de très faible rigidité. Il permet ainsi aux cellules souches neurales d’y pénétrer et de s’y développer en trois dimensions.
L’hydrogel présente également un maillage composé de différents types de fibres, les unes droites et rigides ; les autres courbes et flexibles. Cette diversité permet aux neurones de développer un réseau d’interconnexions à courtes et longues distances telles qu’elles sont observées dans le tissu cérébral.
Ce nouveau biomatériau pourrait donc permettre de développer des modèles de tissu cérébral en trois dimensions dont le fonctionnement se rapprocherait des conditions in vivo. À terme, il pourrait être utilisé pour évaluer l’effet d’un médicament ou permettre la transplantation de cellules avec leur matrice dans le cadre de réparations de lésions cérébrales.
1 Les cellules souches neurales sont issues de biopsies de patients (CHU de Toulouse – Pôle Neurosciences). Ces cellules sont capables de se différencier en neurones et en cellules gliales, les principaux types cellulaires du tissu cérébral.
DOCUMENT inserm LIEN |
| |
|
| |
|
| Page : [ 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 ] Précédente - Suivante |
|
|
| |
|
| |
|